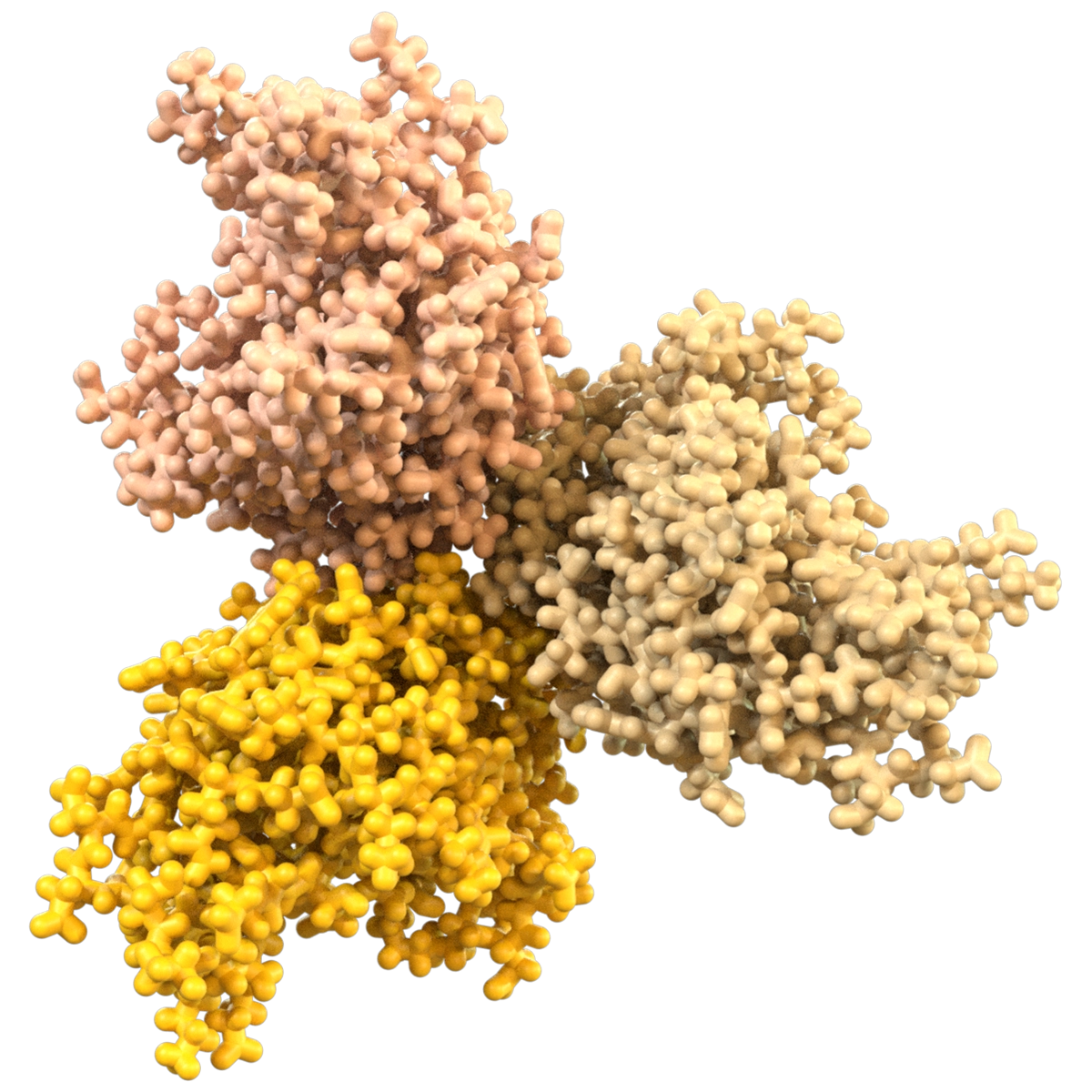

A Moving Target
Proteins are dynamic, shapeshifting molecules. Traditional approaches treat protein targets as rigid structures because predicting protein motion is very challenging. However, modulating biological activity often relies on protein motion.
Doing the Previously Impossible
By predicting the fundamental relationship between protein motion and disease, we can create novel small molecules for targets with little or no known chemical matter—disease targets that previously have been considered “undruggable” with small molecules.